在过去的三年里,FDA批准的新药数量一直处于历史的高位,但今年却出现了大幅减少,截止12月31日,CDER仅批准了37个新药,这些新药中,将会有7-8个产品在上市第五年的销售额超过10亿美元,成为重磅炸弹。本文就以上37个新药进行简单地回顾和综述,希望为广大读者带来便捷和帮助。
1. Daridorexant(商品名:Quviviq)
1月7日,FDA批准了IDORSIA公司的Daridorexant,用于成人失眠症治疗。本品是一种食欲素受体阻滞剂,与此前FDA批准的Suvorexant作用机制相同。
FDA批准本品上市的主要依据是两项三期临床试验的结果,1854名受试者分别接受了本品50mg,本品25mg或安慰剂治疗,试验主要终点为治疗后1个月、3个月的入睡后觉醒时间(WASO)与睡眠潜伏期(LPS)相比基线的变化。
结果显示,在治疗1个月后,50mg组、安慰剂组的WASO分别相比基线缩短29、18和6分钟,LPS分别基线缩短31、28和20分钟,而治疗3个月后,三组的WASO分别相比基线缩短29、23和11分钟,LPS则分别相比基线缩短35、31和23分钟。
两项临床试验说明,经过本品治疗,患者睡眠质量得到显著改善。市场潜力方面,睡眠障碍的患者众多,Ealuatepharma(EVP)预测本品在2026年的销售额可达5.31亿美元。
2. Abrocitinib (商品名:Cibinqo)
1月14日,FDA批准了辉瑞公司的Abrocitinib,用于难治性的中重度特应性皮炎(AD)品是一种可逆性的JAK1抑制剂,三项临床试验评估了本品的安全有效性。
三项临床试验共计纳入了1615名患者,试验主要终点均为12周的研究者整体评分(IGA)为0分或1分的患者比例和湿疹面积及严重程度指数相比基线改善75%以上患者的比例(EASI-75)。
试验一和试验二为单药治疗,试验三为联合用药治疗。
试验一12周治疗结果显示,本品200mg、100mg和安慰剂组IGA为0或1分的患者比例为44%,24%和8%,而试验二分别为38%,28%和9%,试验三则分别为47%,36%和14%。
EASI方面,试验一中三组患者分别有62%,40%和12%的患者达到EASI-75标准,而试验二分别为61%、44%和10%,试验三则分别为68%,58%和27%,三项试验均达到了预设的主要和次要终点。
AD在发达国家儿童中的患病率高10-20%,成年约为1-5%,患者群体巨大。虽然获批上市的药物众多,但真正能用于中重度患者的口服疗法比较有限,EVP预测本品在2026年的销售额可达7.6亿美元。
3. Tebentafusp(商品名:Kimmtrak)
1月25日,FDA批准了IMMUNOCORE公司T细胞受体(TCR)疗法Tebentafusp,用于HLA-A*02:01阳性,且不可切除或转移性葡萄膜黑色素瘤治疗。
本品是一种双特异性 gp100 肽-HLA 介导的CD3 T细胞接合剂,TCR臂可与黑色素瘤细胞的细胞表面的gp100 肽结合,进而发挥治疗作用。
在一项临床试验中,378名符合条件的患者按2:1分组,分别接受本品或标准疗法(PD-1,CALT4或化疗)治疗,中位总生存期(mOS)分别为21.7个月 vs 16.0个月,中位无进展生存期(mPFS)分别为3.3个月 vs 2.9个月,可观应答率为9.1% vs 4.8%。临床试验结果说明,相比现有的疗法,本品可显著降低患者的死亡风险。
4. Faricimab(商品名:Vabysmo)
1月28日,基因泰克公司的Faricimab喜获FDA批准,用于年龄相关的黄斑病变或糖尿病黄斑病变治疗。
本品是一种血管表皮细胞生长因子和血管生成素抑制剂,两项临床试验证实了本品对年龄相关的黄斑病变的安全有效性。
两项试验共计有1329名患者入组,经过长达2年的治疗和随访,试验结果本品的疗效非劣于阿柏西普。糖尿病黄斑病变方面,该公司同样开展了两项临床试验,两项试验共计有1891名患者入组,两项临床试验的结果同样证实,本品的疗效非劣性于阿柏西普。
由于年龄相关的黄斑病变或糖尿病黄斑病变的治疗市场巨大,雷珠单抗、阿柏西普都是家喻户晓的重磅炸弹,本品疗效非劣于阿柏西普,EVP对该产品的预期较高,预测该产品在2026年的销售额可达11.29亿美元。
5. Sutimlimab(商品名:Enjaymo)
2月4日,FDA批准了BIOVERATIV医疗公司的Sutimlimab,用于冷凝集素病(CAD)患者治疗,以降低患者因溶血而对红细胞输注的依赖。
CAD是冷抗体型自身免疫性溶血性贫血,是免疫球蛋白M引起的免疫性疾病,而本品是一种经典补体抑制剂,可与补体C1结合,抑制经典补体途径。
在一项开放标签,单臂设计的临床试验中,24名患者接受了本品治疗,其中13名达到应答标准(血红蛋白浓度超过12 g/dL或血红蛋白浓度相比基线增加2 g/dL以上,在治疗后第5-26周无红细胞输注、无需接受标准疗法治疗)。
其中9名患者血红蛋白浓度超过了12 g/dL,15名相比基线提升了2 g/dL,在5-26周间,17名患者未接受红细胞输注,22名患者未接受CAD标准方案治疗。
CAD是一种极其罕见的疾病,患病率约1/10万-2/10万,本品是首个针对性的疗法。约占自身免疫溶血性贫血患者的15%。虽然潜在患者群极其稀少,但有效的治疗手段不多,EVP预测本品在2026年的销售额可达4.4亿美元。
6. Mitapivat(商品名:Pyrukynd)
2月17日,FDA批准了AGIOS制药的Mitapivat,用于丙酮酸激酶(PK)缺乏所引起的溶血性贫血治疗。
PK缺乏症是一种由丙酮酸激酶基因异常所致的溶血性贫血,而本品是一种PK激动剂,FDA批准本品是基于两项临床试验的研究结果。
其中一项是针对未定期输血的患者(试验开始前52周输血不超过4次,3个月内未接受输血),共有80名患者加入到临床试验,试验的主要终点为第16、20和24周时,血红蛋白浓度相比基线增加1.5g/dL的患者比例。
其结果显示,本品治疗的40名患者,16%的患者出现了应答,而安慰剂组治疗的患者,应答率为0%。
另一项试验是针对定期输血患者设计的开放性试验(试验开始前的52周内至少有6次输血),共有27名患者参与了试验,试验的主要终点为在一定时期内,患者的红细胞输注量相比历史下降33%以上。结果显示,33%的患者达到了应答标准,其中22%的患者未输血。
PK缺乏症是一种罕见性疾病,患病率约为两万分之一,EVP预测该产品在2026年的销售额为2.87亿美元。
7. Pacritinib(商品名:Vonjo)
2月28日,FDA加速批准了CTI生物制药公司的Pacritinib,用于血小板计数低于50×10^9/L的中度或高危原发性或继发性的骨髓纤维化。
骨髓纤维化(MF)是一种费城染色体阴性的骨髓增生性肿瘤,临床表现主要是贫血、脾脏肿大,大部分患者与JAK信号通路传导失调有关。本品是一种JAK2和FLT3抑制剂,在一项311名患者参与的临床试验中,本品的安全有效性得到了初步的确证。
入组患者按1:1:1的比例分别接受本品400mg每日一次、本品200mg每日2次和现有的最佳方案治疗。在该试验中,每日一次400mg未确定安全性,也未获得批准,而在每日两次200mg,且基线血小板计数低于 50 × 10^9的患者队列中获得了有效性数据。
31名患者接受本品每日两次200mg治疗24周,29%的患者脾脏体积相比基线缩小了35%以上,而接受标准疗法的32名患者中,仅有3.1%达到这一标准。市场潜力方面,EVP预测该产品2026年的销售额可达3.68亿美元。
8. Ganaxolone(商品名:Ztalmy)
3月18日,FDA批准了MARINUS公司的ganaxolone,用于细胞周期蛋白依赖性激酶样-5 (CDKL5)缺乏症相关的癫痫治疗。
CDKL5缺乏症是一种严重而罕见的遗传性癫痫,本品的作用机制尚未完全清楚。FDA批准该产品上市,是基于一项有101名患者参与的临床试验,患者按1:1的比例进行分组后,分别接受本品或安慰剂治疗,主要终点是28天的癫痫发作频率相比基线值的变化。经过治疗,安慰剂组和本品治疗组的癫痫发作频率分别相比基线值下降7%和31%,达到了统计学上的显著性差异。
CDKL5缺乏症的患病率约为Dravet综合征的一半,平均4-6万名存活婴儿中,就会出现一名CDKL5缺乏症患者,因为治疗手段有限,EVP预测本品在2026年的销售额可达3.84亿美元。
9. Nivolumab和Relatlimab(商品名:Opdualag)
同在3月18日,FDA也批准了百时美施贵宝公司的新型免疫疗法Opdualag,用于无法手术或转移性黑色素瘤治疗。
本品由PD-1抗体Nivolumab和LAG-3单抗RELATLIMAB组成,其中RELATLIMAB是一种全新的免疫检查点抑制剂。
在一项临床试验中,714名3级或四级无法手术或转移性的黑色素瘤患者,分别随机接受本品(n=355)或Nivolumab单独治疗(n=359),结果显示,mPFS分别为10.1个月 vs 4.6个月,mOS分别为NR vs 34.1个月,可观应答率分别为43% vs 33%。相比单药疗法,本品的疾病控制率大幅提高,死亡风险下降了25%。
虽然免疫检查点抑制剂潜在的适应症较多,但EVP对本品在2026年的销售额预测值仅为4.37亿美元。
10. 镥-177 vipivotide tetraxetan (商品名:Pluvicto)
3月23日,诺华的新型放射型药物喜获FDA批准,用于前列腺特异性膜抗原(PSMA)阳性的去势抵抗的转移性前列腺癌。
本品是一种含镥-177的放射性药物,在一项临床试验中,患者按2:1的比例分组,分别接受本品联合最佳支持治疗(BSoC)或BSoC单独治疗,结果显示接受联合治疗的551名患者,mOS为15.3个月,而接受BSoC单独治疗的280名患者,mOS仅为11.3个月,两组患者的客观缓解率分别为30% vs 2%,达到了预设的主要终点。
前列腺癌是西方发达国家男性发病率最高的恶性肿瘤,患者众多,EVP预测本品在2026年的销售额可达8.51亿美元。
11. Oteseconazole(商品名:Vivjoa)
4月26日,FDA批准了MYCOVIA制药的oteseconazole,用于外阴道念珠菌病复发预防。本品是一种新型抗真菌药,体外试验证实对多种念珠菌均有活性,FDA批准本品上市,是基于三项临床试验的研究结果。
其中试验一和试验二为安慰剂对照的随机试验,试验重点皆为48周内,念珠菌培养确定的急性发作((念珠菌属真菌培养阳性,临床体征和症状评分为≥3))次数≥1次的患者比例。
结果显示,试验一中,本品治疗组和安慰剂组患者的急性发作比例分别为6.7%和42.8%,试验二中,两组患者的急性发作比例分别为3.9%和39.4%,均达到了预设的终点。念珠菌性阴道炎是极为常见的疾病,在20-30岁年龄段妇女中,患病率高达80%,处女和绝经女性患病较低。
尽管如此,但药物较多,复发病例较少,故本品销售额达到重磅炸弹级别的概率较低。
12. Mavacamten(商品名:Camzyos)
4月28日,FDA批准了百时美施贵宝公司的Mavacamten,用于梗阻性肥厚型心肌病治疗,以改善患者的心脏功能和症状。
本品是一种选择性的可逆心肌肌球蛋白抑制剂,FDA批准本品上市是基于一项三期试验的研究结果。
251名纽约心脏病协会(NYHA)分组为II-III级的梗阻性肥厚型心肌病患者分别接受本品或安慰剂治疗,试验终点为30周时评估心脏功能,混合静脉血氧张力改善≥1.5 mL/kg/min外加NYHA分组改善1级以上的患者比例或混合静脉血氧张力改善≥3.0 mL/kg/min外加NYHA分组无恶化的患者比例。
结果显示,接受本品治疗的123名患者中,37%的患者达到了试验终点,而接受安慰剂治疗的128名患者中,仅有22%的患者达到了试验终点。
试验结果说明,本品相比安慰剂,可显著性改善患者的心脏功能。市场方面,梗阻性肥厚型心肌病患病率高达五百分之一,尚未满足的治疗需求较大,EVP预测该产品在2026年的销售额可达16.72亿美元,是2022年获批的第二个潜在重磅炸弹。
13. 阿莫西林、克拉霉素和沃诺拉赞(商品名:Voquezna triple pak)
5月3日,FDA批准了PHATHOM 公司的Voquezna triple pak,用于幽门螺旋杆菌感染治疗。
本品含有β-内酰胺类抗生素阿莫西林,大环内酯类抗生素克拉霉素和新型抗酸药沃诺拉赞,FDA批准本品上市是基于一项三期临床试验的研究结果。
入组患者按1:1:1的比例,分别接受沃诺拉赞20mg每日二次+阿莫西林1000mg每日二次+克拉霉素500mg每日二次(沃诺拉赞三联治疗组),沃诺拉赞20mg每日二次+阿莫西林1000mg每日三次(沃诺拉赞二联治疗组),或兰索拉唑30mg每日两次+阿莫西林1000mg每日二次+克拉霉素500mg每日二次(兰索拉唑赞三联治疗组)治疗,治疗27天后,幽门螺旋杆菌检测阴性为治疗成功。
结果显示,沃诺拉赞三联治疗组治疗成功率为80.8%,沃诺拉赞二联治疗组治疗成功率为77.2%,而兰索拉唑赞三联治疗组仅为68.5%。
虽然是三联疗法,但沃诺拉赞在日本、中国上市已经多年,而且化合物专利期将满,未来五年里,本品销售额超过10亿美元的概率并不大。
14. Tirzepatide(商品名:Mounjaro)
5月13日,FDA批准了礼来公司的Tirzepatide,在饮食控制和运动的基础上,用以控制二型糖尿病患者的血糖。本品是一种GLP-1和GIP双靶点激动剂,为了获得FDA的批准,礼来公司已经开展了多项临床试验研究。
单药治疗方面,经过本品不同剂量的40周治疗,本品5mg、10mg和15mg治疗组的糖化血红蛋白分别相比基线下降了1.8%、1.7%和1.7%,而安慰剂组仅下降了0.1%,三个治疗组患者的平均体重分别相比基线下降了6.3kg、7.0kg和7.8kg,而安慰剂组仅下降了1.0kg。
在二甲双胍为背景治疗的基础上添加本品治疗方面,本品5mg、10mg和15mg治疗组的糖化血红蛋白分别相比基线下降了2.0%、2.2%和2.3%,而索马鲁肽治疗组(对照)则仅下降了1.9%,体重方面,本品三个治疗组分别相比基线下降7.6kg、9.3kg和11.2kg,而索马鲁肽治疗组则仅下降了5.7kg。
在基础胰岛素的背景治疗下,添加本品治疗方面,本品5mg、10mg和15mg治疗组的糖化血红蛋白分别相比基线下降了2.1%、2.4%和2.3%,而安慰剂组仅下降了0.9%,体重方面,本品三个治疗组分别相比基线下降5.4kg、7.5kg和8.8kg,而安慰剂组则上涨了1.6kg。
GLP-1类似物是当今最火热的糖尿病药物,但本品不论是降糖还是减重都明显优于常见的GLP-1类似物,EVP预测本品在2026年的销售额可达54.41亿美元,是一个典型的超级重磅炸弹。
15. Tapinarof(商品名:Vtama)
5月23日,FDA批准了DERMAVANT公司的Tapinarof乳膏,用于斑块银屑病治疗。本品是一种芳香烃受体激动剂,两项安慰剂对照的临床试验证实了本品的安全有效性。
在两项试验中,患者均按2:1的比例分组,分别接受本品和安慰剂治疗,试验终点均为12周时的医师总体评分(PGA)为0分(斑块清除)和1分(几乎清除)的患者比例。
结果显示,两项试验中的本品治疗组分别有36%和40%的患者达到了治疗终点,而安慰剂组则分别只有6%和6%。治疗斑块银屑病的药物众多,而本品并不出众,EVP也并未就本品给出市场预期。
16. Vutrisiran(商品名:Amvuttra)
6月13日,FDA批准了ALNYLAM公司的Vutrisiran,用于遗传性转甲状腺素淀粉样变所致的多发性神经病治疗,本品是一种由GalNAc递送的小干扰RNA,FDA批准本品上市是基于一项开放标签的临床试验结果。
入组患者按3:1的比例分别接受本品或patisiran治疗,主要终点为9个月时患者调整神经病变缺损评分(mNIS+7)相比基线值的变化,结果显示,本品治疗组相比基线下降了2.2分,而安慰剂组则上升了14.8分。
相比使用脂质复合物递送的patisiran,本品并未出现疗效上的改善,但只需要三个月一次皮下注射,患者的顺应性得到大幅提升,EVP预测本品在2028年的销售额可达27.22亿美元。
17. Olipudase alfa (商品名:Xenpozyme)
8月31日,健赞公司的Olipudase alfa获得了FDA的批准,用于酸性鞘磷脂酶缺乏症(ASMD)疗。是一种由酸性鞘磷脂酶缺乏引起的罕见脂质贮积症,主要表现是肝脾肿大,呼吸困难。
本品是一种外源性的酸性鞘磷脂酶,在一项随机、双盲、安慰剂对照的临床试验中,患者分别接受本品或安慰剂治疗52周,主要终点为肺一氧化碳弥散量(DLco)、脾脏体积、肝脏体积、血小板计数相比基线时的变化。
经过52周的治疗,本品治疗组平均DLco相比基线上升了3%,而安慰剂组则上升了23.9%;脾脏体积方面,本品治疗组相比基线增加了0.5%,而安慰剂组则相比基线下降了38.9%;
脾脏体积方面,本品治疗组相比基线下降了1.8%,而安慰剂组则相比基线下降了26.5%;而血小板计数方面,本品治疗组相比基线增加了2.7%,而安慰剂组则相比基线增加了18.3%。
ASMD是一种极其罕见的疾病,患病率为4/100万-6/100万,故显而易见的是本品又将是一个天价药物,EVP预测本品在2028年的销售额为3.46亿美元。
18. Spesolimab(商品名:Spevigo)
9月1日,勃林格殷格翰的Spesolimab获得FDA批准上市,用于全身性脓疱型银屑病治疗。本品是一种IL-36受体抑制剂,FDA批准该产品上市的依据是一项多中心、安慰剂对照、随机双盲试验的研究结果。
53名受试患者分别接受本品(n=35)或安慰剂(n=18)治疗,试验主要终点为治疗一周后的全身性脓疱型银屑病医生总体评分(GPPPGA)为0分(无可见脓疱)的患者比例。结果显示本品治疗组有54%的患者达到的预设的试验终点,而安慰剂组仅为6%。
19. Daxibotulinumtoxin A(商品名:Daxxify)
9月7日,FDA批准了REVANCE医疗公司的Daxibotulinumtoxin A,用于暂时性改善中重度眉间皱纹的外观。本品是一种新型肉毒杆菌毒素,是一种乙酰胆碱释放抑制剂,FDA批准本品上市是基于两项临床研究的结果。
合并两项试验,共有406名患者接受了本品治疗,剩余的203名患者则被给予安慰剂。两项试验通过研究者和受试者的评分量表来确定眉间皱纹的严重程度,0分为无皱纹,1分轻度,2分为重度,3分为重度,试验终点均为4周后评分为0分、1分或相比基线改善两分以上的患者比例。
两项试验中,本品治疗组均有74%的患者达到了治疗终点,而安慰剂组均为0%。在本品获批之前,美国已经有三种不同的肉毒杆菌毒素上市销售,市场潜力方面,EVP预测本品在2028年的销售额为5.23亿美元。
20. Deucravacitinib(商品名:Sotyktu)
9月9日,百时美施贵宝公司的Deucravacitinib喜获FDA批准上市,用于中重度斑块银屑病治疗。本品是一种酪氨酸激酶2(TYK2,JAK的家族一员)抑制剂,两项临床试验证实了本品的安全有效性。
两项试验共有1684名患者入组,分别接受本品、阿普斯特或安慰剂治疗,试验终点为16周时,PGA评分为0分或1分的患者比例及PASI 75达标患者比例。
在试验一中,本品、阿普斯特或安慰剂治疗组16周时PGA评分为0分或1分的患者比例分别为54%、32%和7%,而PASI 75达标患者比例分别为58%、35%和13%。
试验二中,三组16周时PGA评分为0分或1分的患者比例分别为50%、34%和9%,而PASI 75达标患者比例分别为53%、40%和9%。阿普斯特是中重度斑块银屑病治疗方面的重磅炸弹,本品疗效比阿普斯特更为出色,EVP预测本品在2028年的销售额可达16亿美元。
21. Eflapegrastim(商品名:Rolvedon)
同在9月9日,SPECTRUM公司的Eflapegrastim也获得了FDA批准,用于非髓性恶性肿瘤患者接受骨髓抑制性抗癌药物时所致的中性粒细胞减少症治疗,以降低感染发生率。
本品是一种重组人粒细胞集落刺激因子的融合蛋白,是一种超长效注射剂,两项与培非格司亭头对头的临床试验证实了本品的安全有效性。两项试验共计有643名接受多西他赛和环磷酰胺化疗的早期乳腺癌患者参与了临床试验,试验终点均为第一个疗程中严重中性粒细胞减少的持续时间(DSN)。
在试验一中,本品治疗的196名患者,DSN的平均值和中位值分别为0.20天和0天,而培非格司亭治疗的210名患者,DSN的平均值和中位值分别为0.35天和0天。
试验二中本品治疗的118名患者,DSN的平均值和中位值分别为0.31天和0天,而培非格司亭治疗的119名患者,DSN的平均值和中位值分别为0.39天和0天。两项试验均说明本品非劣性与培非格司亭。市场潜力方面,EVP预测本品在2028年的销售额可达2.97亿美元。
22. 特利加压素(商品名:Terlivaz)
9月14日,FDA批准了马林克罗特公司的特利加压素,用于改善肾功能迅速下降的肝肾综合征患者的肾功能。本品是一种加压素,在我国已经上市多年,FDA批准本品在美国上市的主要依据是一项多中心临床试验的研究结果。
该项临床试验一共招募了300名肾功能快速恶化的肝硬化、腹水、1型肝肾综合症患者,所有入组患者均为血清肌酐(SCr)≥2.25 mg/dL,两周内SCr值翻倍、利尿剂停药后48小时内肾功能无持续改善和开始使用白蛋白进行血浆扩张的患者。
入组患者按2:1分组分别接受本品或安慰剂治疗,试验终点为肝肾综合征的逆转率(治疗14天或出院时,两个小时内连续两次SCr检测值小于1.5 mg/dL)。
结果显示,本品治疗组有29.1%的患者达到了治疗终点,而安慰剂组仅为15.8%。本品治疗组有31.7%的患者肝肾综合症持续逆转至第30个治疗日,而安慰剂组仅为15.8%。
23. Gadopiclenol(商品名:Elucirem)
9月21日,FDA批准了GUERBET公司的新型含钆造影剂Gadopiclenol,用于中枢神经系统和身体各部位的核磁显影。
两项临床试验证实了本品的安全有效性,其中试验一有256名患者入组,是针对中枢神经系统病变显影的临床试验,试验二有309名患者入组,是针对身体病变显影的临床试验。
两项试验中,患者均使用用了本品,通过对比使用造影剂前后的核磁成像结果来反映本品的显影效果。
三位读片人分别对各个患者病灶的边界描绘、内部形态和对比度增强进行一一评分,并确定病灶数量。两项试验结果均证实本品治疗组的病灶可视化评分、确定病灶数量均与钆布醇相似。
24. Omidenepag isopropyl(商品名:Omlonti)
9月22日,FDA批准了参天制药的Omidenepag isopropyl 滴眼液,用于降低开角型青光眼所致的眼压升高。
本品是一种前列腺素E2受体激动剂,三项临床试验证明了本品的安全有效性。
在三项试验中,入组患者的基线眼压在24-26 mm汞柱,经过三个月的治疗,本品治疗组患者眼压下降了5-7mm汞柱,对应的噻马洛尔组也5-7mm汞柱,而拉坦前列素治疗组则下降了6-8 mm汞柱。
本品的获批上市意为着,人工合成的前列腺素受体激动剂也可以达到前列素类似物相同的效果,青光眼有望摆脱对前列腺素的依赖。
25. 牛磺酸二醇和苯丁酸钠(商品名:Relyvrio)
9月29日,FDA批准了AMYLYX公司的牛磺酸二醇和苯丁酸钠组合物,用于肌萎缩侧索硬化治疗。
虽然本品的作用机制尚不清楚,但一项有137名患者参与的临床试验证明了该产品的疗效。入组患者按2:1进行分组,分别接受本品或安慰剂治疗24周,主要终点是24周时,意向治疗人群ALSFRS-R(修订ALS功能评分量表)评分的差异。
结果显示,本品治疗组为29.06分,而安慰剂组为26.73分,达到了统计学显著性差异。虽然两组患者评分都有明显下降,但经过本品治疗,下降速度有所减缓。
26. Futibatinib(商品名:Lytgobi)
9月30日,FDA加速批准了TAIHO肿瘤治疗公司的Futibatinib,用于成纤维细胞生长因子受体2(FGFR2)基因融合或其他重排,且既往接受过治疗、无法手术的局部进展或转移性肝内胆管癌治疗。
本品是一种FGFR抑制剂,可抑制f FGFR 1、f FGFR 2、f FGFR 3和f FGFR 4。在一项有103名患者参与的临床试验中,经过本品的开放性治疗,客观缓解率为42%,mDoR为9.7个月。
胆管癌比较少见,美国每年新确诊患者约为8 000 人,其中15% 为FGFR2 基因突变。市场潜力方面,EVP预测本品在2028年的销售额为0.8亿美元。

27. Tremelimumab(商品名:Imjudo)
10月21日,FDA批准了阿斯利康的CTLA-4抗体tremelimumab,与该公司此前已经获批的PD-L1抗体Imfinzi(durvalumab)联用,治疗无法手术的肝细胞癌。
临床试验数据显示,本品与Imfinzi联合治疗的393名患者,mOS为16.4个月,而索拉非尼的389名患者为13.8个月,死亡风险下降了22%。
客观缓解率方面,本品与Imfinzi联合治疗组为20.1%,而索拉非尼组仅为5.1%,联合治疗组有3.1%的患者完全缓解,而索拉非尼组仅为0%。
CTLA-4与PD-1/PD-L1联合使用具有一定的协同作用,此前FDA也批准了百时美施贵宝的Opdivo联合Yervoy治疗无法手术的肝细胞癌,客观缓解率为33%,但临床终点数据尚未成熟,而且试验样本量也比较小。
相比BMS的CTLA-4与PD-1联合治疗,默沙东的PD-1单药(Keytruda)治疗,客观缓解率就稍微低一些,仅有17%,该产品的临床终点数据也尚未成熟。
肝癌HCC是较为常见的恶性实体瘤,80%以上集中在发展中国家,发达国家虽患病率较低,但近年来有明显的上升趋势,据文献报道,全球每年约有56万新发患者,由于肝癌信号通路较为复杂,近年来虽然研究很热,但并未带来明显的突破,肝癌仍是预后最差的常见癌症之一。
28. Teclistamab (商品名:Tecvayli)
美国FDA于10月25日加速批准了强生的双特异性抗体Teclistamab,用于复发或难治性的多发性骨髓瘤四线治疗。
Teclistamab是一种可皮下注射的抗体,维持期治疗只需每周注射一次,这相比静脉注射的产品是一种优势。
临床试验显示,参与试验的110名受试患者中68名患者产生了应答,客观应答率为61.8%,其中31名患者完全缓解,完全缓解率为28.2%。
由于是加速批准,该产品的临床试验终点尚未成熟。据文献报道,全球约有一万名多发性骨髓瘤四线患者,但目前获批药物较多,本品脱颖而出的关键除了疗效优势外,还有一二三线疗法的开拓。市场潜力方面,EVP预测本品在2028年的销售额可达17.04亿美元。
29. Mirvetuximab soravtansine(商品名:Elahere)
11月14日,FDA加速了批准ImmunoGen公司研发的Mirvetuximab soravtansine用于叶酸受体α(FRα)阳性,铂耐药上皮性卵巢癌、输卵管癌或原发性腹膜癌的成年患者,这些患者之前接受过一至三种全身治疗方案。
ELAHERE是一种抗体药物偶联物(ADC),Mirvetuximab 是一种IgG1,可靶向于叶酸受体α,而小分子部分是一种微管抑制剂,该产品只需三周注射一次。
在一项有104名铂耐药的卵巢癌患者参与的临床试验中,总缓解率为31.7%,其中完全缓解率为4.8%,部分缓解率为26.9%,中位持续缓解时间(mDoR)是6.9个月,临床终点数据尚未成熟。市场潜力方面,EVP预测本品2028年的销售额可达7.59亿美元。
30. Teplizumab(商品名:Tzield)
11月17日,美国食品药品管理局批准了teplizumab,用于以延缓1型糖尿病的发病期,可延缓3期的1型糖尿病成年人和10岁以上的2期儿童的发作时间。
1型糖尿病是一种免疫系统攻击并破坏胰岛细胞的疾病,约占糖尿病患者总数的5-10%,1型糖尿病需要注射胰岛素维持生存。
本品可以使攻击胰岛的免疫细胞失活,同时增加有助于调节免疫应答的细胞比例。在一项有76名2期1型糖尿病患者参与的随机、双盲、安慰剂对照试验中,患者分别每天一次接受安慰剂或每14天一次接受本品治疗。主要终点是3期1型糖尿病的诊断时间。
在51个月的中位随访中,接受Tzield治疗的44名患者诊断率为44%,而接受安慰剂治疗的32名患者诊出率为72%,接受Tzield治疗的患者组,中位诊出时间为50个月,而安慰剂组为25个月,数据证明3期1型糖尿病的发展时间显著性延迟。市场方面,EVP预测本品在2028年的销售额可达11.71亿美元。
31. Olutasidenib(商品名:Rezlidhia)
12月1日,FDA批准了Forma 医疗公司的olutasidenib,用于复发或难治性异柠檬酸脱氢酶1(IDH1)阳性的急性粒细胞白血病(AML)治疗。
在多种实体瘤和非实体瘤患者中,异柠檬酸脱氢酶阳性突变较为常见,在AML患者中,15-20%的患者可见异柠檬酸脱氢酶阳性。
在美国,AML的发病率为4.3/10万,2019年的新发病例为2.15万人,IDH1阳性的AML新发患者约为4000人。本品是一种新型IDH1抑制剂,其安全有效性在一项2期开放标签的临床试验中得到初步评估,153符合入组条件的患者参与了临床试验,其中123名患者获得了有效性数据。
结果显示,33%的患者出现了完全缓解或部分血液学缓解,在获得缓解的患者中,18个月的存活率为87%。中位缓解时间、无进展生存期、总生存期等数据尚未达到。除了本品,此前FDA批准的同靶点产品还有Enasidenib和Ivosidenib。
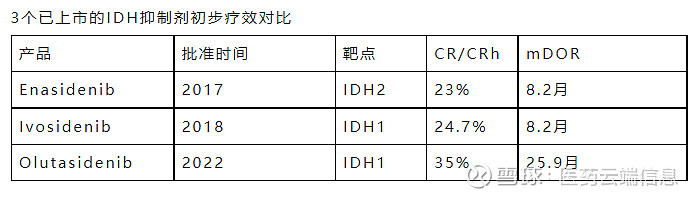
32. Adagrasib(商品名:KrazatiI)
12月12日,FDA加速批准了MIRATI医疗公司的Adagrasib,用于KRAS G12C基因突变的局部进展或转移性非小细胞肺癌(NSCLC)治疗。
本品是一种不可逆的KRAS G12C抑制剂,在一项多中心、单臂设计,开放标签的临床试验中,112名符合条件的患者接受本品治疗后,客观缓解率为43%,mDoR为8.5个月,PFS、OS等临床终点尚未达到。
KRAS G12C突变是NSCLC较为常见的突变类型,约占患者总数13%。在此之前,KRAS 是长期被认为难以成药的靶点,2021年5月,安进的Sotorasib,开启了该靶点治疗的新篇章,除了已经获批的两个药物,目前还有多个产品处于研发阶段。市场方面,EVP预测本品在2028年的销售额可达20亿美元。
33. Lenacapavir(商品名:Sunlenca)
12月22日,FDA批准了吉利德的超长效衣壳蛋白抑制剂Lenacapavir,在联合其它抗逆转录药物的基础上,用于治疗高度耐药的HIV-1感染患者,包括现有的治疗方案因耐药治疗失败、不耐受或因安全性考虑而无法使用现有治疗方案治疗的患者。
本品是一种全新机制的抗艾滋病药物,特点是半衰期长,口服给药的半衰期长达10-12天,而大剂量注射剂的半衰期长达8-12周,故只需半年注射一次。
本品的安全有效性,在一项为期52周,安慰剂对照,随机双盲的多中心临床试验中得到了初步的确认。
72名既往经接受多种方案治疗失败,且体内病毒载量≥ 400拷贝/ml的患者被分为两个队列,36名在随访中病毒载量下降量小于0.5 log10拷贝/ml的患者进入队列一(随机组),而下降量大于等于0.5 log10拷贝/ml或超过队列一样本数量的患者被纳入队列二(非随机组)。
队列一入组后,按2:1的比例随机接受本品或安慰剂进行为期14天的功能性治疗,功能性治疗结束后再接受最佳治疗方案+本品联合治疗。
主要治疗终点为在功能性治疗期间,体内病毒载量下降大于等于0.5 log10拷贝/ml的患者比例。
结果显示,本品治疗组有87.5%的患者达到了治疗终点,而安慰剂组仅为16.5%。
在后续的长期治疗中,26周和52周体内病毒载量低于50拷贝/ml的患者比例分别为81%和83%。队列二的患者则入组后直接接受最佳治疗方案+本品联合治疗,26周和52周体内病毒载量低于50拷贝/ml的患者比例分别为81%和72%。
在此之前,EMA已经于8月份率先批准了本品上市,市场潜力方面,EVP预测本品在2026年和2028年的销售额分别为8.77亿美元和16.02亿美元。
34. Mosunetuzumab(商品名:Lunsumio)
12月22日,FDA加速批准了基因泰克公司的Mosunetuzumab,用于复发或难治性滤泡淋巴瘤(FL)三线治疗。
本品是一种双特异性抗体,可同时靶向作用于T细胞表面的CD3和淋巴瘤细胞表面的CD20抗原。
该产品的安全有效性在一项多中心,开放标签的临床试验中得到了评估。既往经历过二线治疗(含CD20抗体)的90名患者在接受本品治疗后,ORR为72%,mDoR为22.8个月,58%的患者持续缓解时间超过了18个月。市场潜力方面,EVP预测本品在2026年的销售额可达5.25亿美元。
35. 超极化氙-129(Xenoview)
12月23日,FDA批准了POLAREAN公司的超极化氙-129吸入剂,用于核磁成像,辅助患者的肺部通气情况评估。
两项试验证明了本品的安全有效性,入组患者分别接受本品或氙-133显影,然后分别评估呼吸道各个区域的信号评分,两项试验均证实,本品和氙-133成像之间的差异在预先指定的等效范围内,观察估值分别为1.4%和1.6%。
36. Ublituximab(商品名:Briumvi)
12月28日,FDA批准了TG医疗公司的Ublituximab,用于复发型的多发性硬化症(MS)治疗,包括临床孤立综合征、复发缓解型疾病和活动性继发性进展性疾病。
本品是一种CD20抗体,FDA批准本品上市是基于两项临床研究的结果。在两项试验中,患者(1:1)分别接受本品或特立氟胺治疗96周,入组患者均在入组前的1年中至少复发1次、前2年中至少复发2次、或在前1年中新出现钆(Gd)增强下的可见病灶,扩展的残疾状态量表(EDSS)均处于0-5.5分。两项试验的主要终点均为年后复发率(ARR),
结果显示,本品治疗组在两项试验中的ARR分别为0.076和0.091,而对应的特立氟胺治疗组分别为0.188和0.176,两项试验均达到了主要终点。
37. Anacaulase(商品名:NexoBrid)
同在12月28日,FDA还批准了MEDIWOUND公司的anacaulase,用于去除部分皮层和/或全层深度热烧伤成人的焦痂。
本品是一种植物药产品,含有蛋白水解酶的成分,可用于移除烧伤患者身上的焦痂,此前已经在日本、欧洲的多个国家获批上市。
此次获得FDA批准,主要依据是一项代号为DETECT的临床试验,该试验评估了本品对全身表面积(TBSA)3%-30%的深部和全层烧伤患者的安全有效性。本品相比与空白基质的焦痂清除率≥95%,达到了其主要终点。